
Sars-CoV-2-Antigenschnelltests wegen mangelnder Sensitivität in der Kritik
Who we areNewsPress & MediaNewslettersNewsletter „Healthcare“ArchivNewsletter „Healthcare“ 1/2022
Sars-CoV-2-Antigenschnelltests in der Kritik
Überblick über die regulatorischen Anforderungen der Verordnung über In-vitro-Diagnostika 2017/746 (IVDR) am Beispiel der Sars-CoV-2-Antigenschnelltests
Zwei Jahre nach Beginn der Coronapandemie befinden sich die Infektionszahlen auf einem Hochpunkt. Im Februar 2022 lagen die täglichen Infektionszahlen in Deutschland bei bis zu 248.838 Neuinfektionen pro Tag. Omikron ist in aller Munde und die Welt hofft auf das Ende der Pandemie.
Mittels Antigenschnelltests wird versucht, Covid- 19-Infektionen frühzeitig zu erkennen und so Infektionsketten zu unterbrechen. In diesem Zusammenhang gibt es immer wieder Meldungen über falsch negative und falsch positive Sars-CoV-2-Antigenschnelltests. Besonderes erstere verfehlen das eigentliche Ziel der Tests, sich im Falle einer Infektion in die erforderliche Absonderung zu begeben und die Erreger nicht weiterzugeben. Das Paul-Ehrlich- Institut (PEI) hat daher eine die Sensitivität der Tests vergleichende Liste erstellt, die auch auf der Seite des Bundesinstituts für Arzneimittel und Medizinprodukte (BfArM) abrufbar ist und eine Orientierung im Dschungel der über 600 verschiedenen Schnelltestsysteme geben soll.
Die falschen Testergebnisse geben Anlass zur Frage, welche regulatorischen Anforderungen für Hersteller von Antigenschnelltests gelten. Sars-CoV-2-Antigenschnelltests sind In-vitro-Diagnostika (IVD). Für diese gilt ab dem 26. Mai 2022 zwingend die neue Verordnung 2017/746 über In-vitro-Diagnostika (IVDR), die die bisher geltende Richtlinie über In-vitro- Diagnostika 98/79/EG (IVDD) ablöst und den Anspruch hat, höhere Standards für die Qualität und Sicherheit der Produkte festzulegen.
Dieser Beitrag soll einen kursorischen Überblick über die grundlegenden (Neu-)Regelungen der IVDR am Beispiel von Sars-CoV-2-Antigenschnelltests geben und die Frage beantworten, ob solche Testsysteme mit schlechter Sensitivität unter der IVDR überhaupt noch in den Verkehr gebracht werden dürfen.
Größte Änderung der IVDR: grundlegende Neuklassifizierung von IVD
Mit der IVDR wird die Klassifizierung der Risikoklassen völlig neu gestaltet.
Bislang unterscheidet die IVDD zwischen vier Typen von IVD, namentlich hochkritischen IVD (Liste A), kritischen IVD (Liste B), IVD zur Eigenanwendung sowie sonstigen IVD. Bei den in Liste A und B genannten IVD handelt es sich bspw. um IVD zur Feststellung von Infektionskrankheiten wie Aids, Hepatitis B, C und D, Cytomegalovirus, Chlamydien oder um IVD zur Feststellung von Parametern mit einem hohen individuellen Krankheitsrisiko wie der Tumormarker PSA oder Trisomie 21. Produkte zur Eigenanwendung zeichnen sich dadurch aus, dass sie nach der vom Hersteller festgelegten Zweckbestimmung von Laien in der häuslichen Umgebung angewendet werden können. Unterfällt ein Produkt der Liste A oder B bzw. handelt es sich um ein Produkt zur Eigenanwendung, ist bereits nach der derzeitigen Rechtslage zwingend die Beteiligung einer Benannten Stelle im Rahmen des Konformitätsbewertungsverfahrens erforderlich. Während derzeit auf dem Markt verfügbare Sars-CoV-2-Antigenschnelltests zur Eigenanwendung zwingend der Einbeziehung einer Benannten Stelle bedürfen, können Sars-CoV-2-Antigenschnelltests zum professionellen Gebrauch (z. B. in Testcentern) nach einer einfachen Selbstzertifizierung der Hersteller rechtmäßig in den Verkehr gebracht werden, da sie als „sonstige IVD“ gelten.
Ab dem 26. Mai 2022 werden IVD in die vier Klassen A bis D eingeteilt. Entscheidend sind die Zweckbestimmung der Produkte und die mit ihr verbundenen Risiken. Die Klassifizierung erfolgt anhand von 7 Klassifizierungsregeln. Für die Risikoeinstufung maßgeblich ist eine Kombination aus dem individuellen und öffentlichen Risiko des mittels IVD diagnostizierten Parameters. Je höher das Risiko, desto höher die Risikoklasse.

Fortan haben Hersteller von Sars-CoV-2-Antigenschnelltests im Hinblick auf die Formulierung der Zweckbestimmung, das heißt ob zur Eigenanwendung oder zum professionellen Gebrauch bestimmt, keinen Spielraum im Hinblick auf die Vermeidung der Zusammenarbeit mit einer Benannten Stelle mehr.
Bei den Sars-CoV-2-Antigenschnelltests für den Hausgebrauch handelt es sich weiterhin um Produkte zur Eigenanwendung. Produkte zur Eigenanwendung werden, bis auf einige hier nicht einschlägige Ausnahmen, zukünftig stets der Risikoklasse C zugeordnet.
Sars-CoV-2-Antigenschelltests zum professionellen Gebrauch sowie PCR-Tests hingegen werden stets in die Risikoklasse D eingestuft. Sars-CoV-2-Antigenschnelltests zum professionellen Gebrauch gelten unter der IVDR zudem als sogenannte patientennahe Tests, die nicht für die Eigenanwendung, aber für die Anwendung außerhalb einer Laborumgebung, in der Regel in der Nähe des Patienten oder beim Patienten, durch einen Angehörigen der Gesundheitsberufe bestimmt sind. Patientennahe Schnelltests sind eine namentlich neu benannte Kategorie von IVD, die mit der IVDR eingeführt wurden.
Fachexperten gehen davon aus, dass künftig 90 % der IVD-Hersteller in die Risikoklassen B bis D fallen und damit der Überwachung durch eine Benannte Stelle unterstehen – unter der IVDD waren nur rund 10 % der IVD unter der Aufsicht einer Benannten Stelle.
Gesteigerte Anforderungen an das Konformitätsbewertungsverfahren
Das Konformitätsbewertungsverfahren hat zum Ziel, die Einhaltung allgemeiner Sicherheits- und Leistungsanforderungen nachzuweisen. In Abhängigkeit von der jeweiligen Risikoklasse müssen Hersteller ein bestimmtes Konformitätsbewertungsverfahren durchführen.
Für alle Hersteller von IVD gilt, dass ihre Produkte neben der Erfüllung allgemeiner Sicherheits- und Leistungsanforderungen eine Technische Dokumentation (TD) für das Produkt sowie eine TD für die Überwachung nach dem Inverkehrbringen vorweisen müssen. Während Hersteller von IVD der Klasse A berechtigt sind, bei Erfüllung dieser Anforderungen die Konformitätserklärung selbstständig auszustellen, müssen IVD der höheren Risikoklassen im Rahmen des Konformitätsbewertungsverfahren zusätzliche Anforderungen erfüllen. Alle Hersteller von IVD der Risikoklasse B bis D müssen entweder ein vollständiges Qualitätssicherungssystem vorlegen, wobei es von der Risikoklasse abhängt, ob die TD pro Produktkategorie (Klasse B), pro Produktgruppe (Klasse C) oder pro Produkt (Klasse D) zu erstellen ist. Als Alternative können die Hersteller von Produkten der Klasse C und D das Konformitätsbewertungsverfahren in Form der Baumusterprüfung in Kombination mit Qualitätssicherung für die Produktion wählen.
Die Wahl des Konformitätsbewertungsverfahrens sollte anhand des Update- und Änderungszyklus des IVD erfolgen. Wird ein IVD voraussichtlich gar nicht oder sehr selten über seinen Produktzyklus hinweg geändert, kann die Baumusterprüfung in Kombination mit der Qualitätssicherung der Produktion eine gute Wahl sein. Bei schon absehbaren Änderungen eines IVD ist das Konformitätsbewertungsverfahren durch Einführung eines Qualitätsmanagementsystems und Prüfung der TD zu empfehlen.
Vor allem an die vorzulegende TD stellt die IVDR höhere Anforderungen als die bisher geltende IVDD. Die TD ist Dreh- und Angelpunkt des Konformitätsbewertungsverfahrens, denn sie ermöglicht eine Bewertung der Konformität des IVD anhand der Anforderungen der IVDR und ist zukünftig ausdrücklich in klarer, organisierter, leicht durchsuchbarer und eindeutiger Form zu verfassen. Die TD muss detaillierter sein und jetzt 20 anstatt bisher 13 Anforderungen enthalten. Darüber hinaus wird fortan eine TD über die Überwachung nach dem Inverkehrbringen gefordert, sogenannte Post-Market-Surveillance (PMS). Bei Produkten zur Eigenanwendung, zu denen auch die Sars-CoV-2-Antigenschnelltests zählen, gelten im Hinblick auf die TD zusätzliche Anforderungen, die der Anwendung durch medizinische Laien Rechnung tragen sollen. Herstellern von IVD ist dringend zu empfehlen, ihre bisherige TD einer detaillierten und systematischen Gap-Analyse zu unterziehen, indem die Anforderungen der Anhänge II und III mit den vorhandenen Nachweisdokumenten abgeglichen werden.
Die IVDR hat insbesondere auch die Anforderungen an die in der TD enthaltende Leistungsbewertung erhöht. IVD-Hersteller müssen zukünftig kontinuierlich (!) eine klinische Leistungsbewertung durchführen, um die Einhaltung der grundlegenden Sicherheitsund Leistungsanforderungen nachzuweisen. Grundlage hierfür ist die Erstellung, Dokumentation und fortlaufende Aktualisierung eines entsprechenden Leistungsbewertungsplans. Die Leistungsbewertung hat nach einem genau definierten und methodisch soliden Verfahren zum klinischen Nachweis der drei Säulen wissenschaftliche Validität, Analyseleistung und klinische Leistung zu erfolgen. Mit dem klinischen Nachweis muss wissenschaftlich bewiesen werden, dass der beabsichtigte klinische Nutzen erreicht wird und das Produkt sicher ist. Der Nachweis der klinischen Leistung des IVD muss auf einer klinischen Leistungsstudie, wissenschaftlicher Literatur, die einem Peer-Review unterzogen wurde, oder aus diagnostischen Routinetests gewonnenen veröffentlichten Erfahrungen beruhen. Auf eine klinische Leistungsstudie kann nur dann ausnahmsweise verzichtet werden, wenn es hinreichende Gründe dafür gibt, auf andere Quellen klinischer Leistungsdaten zurückzugreifen. In Bezug auf Sars-CoV-2-Antigenschnelltests muss der IVD-Hersteller also nachweisen, dass der Analyt des Tests im Hinblick auf Sensitivität und Spezifizität geeignet ist, den Sars-Cov-2-Virus möglichst fehlerfrei zu identifizieren, das heißt ohne falsch positive oder falsch negative Ergebnisse. Die Sensitivität gibt an, wie viele der infizierten Personen durch das Testsystem tatsächlich erkannt werden. Eine Sensitivität von 80 % bedeutet, dass 80 von 100 Coronainfizierten ein positives und 20 ein falsch negatives Ergebnis erhalten. Je höher die Sensitivität eines Tests ist, desto sicherer erfasst er die Sars-CoV-2-Infektion.

Unter der IVDD gibt es keine Sensitivitätsanforderungen für Sars-CoV-2-Antigenschnelltests. Das PEI hat mithilfe eines Bewertungspanels des Robert- Koch-Instituts den derzeitigen Stand der Technik für Sars-CoV-2-Antigenschnelltests mit einer Mindestsensitivität von 75 % für die Pools mit einem CT ≤ 25 festgelegt. Sars-CoV-2-Antigenschnelltests, die das Sensitivitätskriterium von 75 % nicht erfüllen, werden als nicht dem Stand der Technik entsprechend und damit unzureichend erachtet. Testsysteme mit einer Sensitivität unter 75 % mit einem CT ≤ 25 wurden daher von der BfArM-Liste über verfügbare Sars-CoV-2-Antigenschnelltests gestrichen.
Nach dem Leitfaden der Medical Device Coordination Group (MDCG) „Guidance on performance evaluation of SARS-CoV-2 in vitro diagnostic medical devices“ soll ein Sars-CoV-2-Antigenschnelltest im Hinblick auf die Analyseleistung unter der IVDR bestimmte quantitative Anforderungen an Sensitivität und Spezifizität erfüllen. Gefordert werden eine Sensitivität von > 80 % und eine Spezifizität von > 98 %. Damit gibt es zukünftig feste Vorgaben für die Mindestsensitivität von Sars-CoV-2-Antigenschnelltests.
Die MDCG ist ein der von der IVDR gefordertes Expertengremium, das Leitfäden erlässt, die nicht rechtsverbindlich sind. Jedoch orientieren sich die Benannten Stellen in der Praxis regelmäßig an den MDCG-Dokumenten und betrachten sie als Stand der Technik. Auch der EuGH bezieht sich in seinen Entscheidungen auf die Leitfäden der MDCG. Benannte Stellen werden damit zukünftig konkrete Anforderungen an Sars-CoV-2-Antigenschnelltests stellen, indem sie eine Mindestsensitivität von 80 % verlangen. Es steht daher zu erwarten, dass sich unter der IVDR die Anzahl solcher Sars-CoV-2-Antigenschnelltests, die vermehrt falsch negative Ergebnisse liefern, reduziert, da diese das Konformitätsbewertungsverfahren nicht erfolgreich durchlaufen.
Darüber ist für IVD der Risikoklasse D, zu denen die Sars-CoV-2-Antigenschnelltests zum professionellen Gebrauch zählen, im Rahmen des Konformitätsbewertungsverfahrens die Hinzuziehung eines EU-Referenzlabors durch die Benannte Stelle verpflichtend. Das EU-Referenzlabor überprüft stichprobenartig, ob das Produkt die vom Hersteller angegebene Leistung auch tatsächlich erbringt, vor allem auch die analytische und diagnostische Sensitivität. Das PEI beabsichtigt, sich bei der Europäischen Kommission für die Benennung als EU-Referenzlabor zu bewerben. Als weitere Maßnahme zur Steigerung der Qualität und Sicherheit von IVD der Klasse D ist bei neuartigen IVD zudem das sogenannte Scrutiny-Verfahren vorgesehen. Die Benannten Stellen sind verpflichtet, ein Expertengremium aus Fachberatern zur Beurteilung der Frage hinzuziehen, ob der Nachweis der Leistung des IVD tatsächlich erbracht wurde, indem sie die Berichte über die Begutachtung der klinischen Bewertung kontrollieren.

Person Responsible for Regulatory Compliance zwingend vorzuhalten
Eine weitere Neuerung ist, dass jeder IVD-Hersteller mindestens eine „Verantwortliche Person“ (Person Responsible for Regulatory Compliance/PRRC) benennen muss, die für die Einhaltung der IVDR-Vorschriften verantwortlich ist. Kleinst- und Kleinunternehmen ist es unter bestimmten Voraussetzungen (< 10 Mio. Jahresumsatz und < 50 Mitarbeiter) gestattet, die PRRC-Stelle auch unter Hinzuziehung eines externen Dienstleisters zu besetzen.
Die Funktionen der PRRC gehen über die des bisherigen Sicherheitsbeauftragten hinaus und der PRRC kommt eine Schlüsselrolle in der Organisationsstruktur des IVD-Herstellers zu: Sie ist unter anderem dafür verantwortlich, die Konformität der IVD in Übereinstimmung mit dem Qualitätsmanagementsystems zu prüfen, die TD aktuell zu halten, die Marktüberwachung durchzuführen sowie die Meldepflichten der IVDR zu erfüllen.
Die PRRC muss über eine festgelegte fachliche Qualifikation verfügen und zudem über mindestens vier Jahre Berufserfahrung in Regulierungsfragen oder mit Qualitätsmanagementsystemen im Zusammenhang mit IVD verfügen. Bei Nichtbenennung einer PRRC droht eine empfindliche Ordnungsstrafe von 30.000 €.
Verlängerte Übergangsfristen
Berechnungen ergeben, dass ab dem 26. Mai 2022 schätzungsweise 50.000 IVD eine Konformitätsbewertung durch eine Benannte Stelle erfordern. Derzeit sind jedoch EU-weit erst sechs Benannte Stellen designiert, darunter die deutschen Unternehmen TÜV Rheinland, TÜV Süd und DEKRA. Man muss kein Rechenkünstler sein, um zu erahnen, dass es den Benannten Stellen nicht möglich sein wird, bis Mai 2022 alle IVD einem Konformitätsbewertungsverfahren zu unterziehen. Die Engpässe der Benannten Stellen sowie die Coronapandemie im Blick, hat die EU Ende 2021 die Übergangsfristen für solche IVD verlängert, die bis zum 26. Mai 2022 auf Grundlage der Richtlinie 98/79 mittels Selbstzertifizierung in den Verkehr gebracht wurden, ab dem 26. Mai 2022 aufgrund der Höherklassifizierung aber einer Benannten Stellen bedürfen. IVD der Klasse D haben drei Jahre länger Zeit, um das Konformitätsbewertungsverfahren durchzuführen. IVD der Klasse C, zu denen auch Sars-CoV-2-Antigenschnelltests zählen, bedürfen erst ab dem 26. Mai 2026 der Bescheinigung einer Benannten Stellen. IVD der Klasse B dürfen sogar noch bis zum 25. Mai 2027 ohne Bescheinigung einer Benannten Stelle in Verkehr gebracht werden. Auch für solche IVD, die bereits unter der Richtlinie 98/79 einer Benannten Stelle bedurften, wurde der Geltungsbeginn der IVDR vom 26. Mai 2024 um ein Jahr auf den 26. Mai 2025 verlängert. Darüber hinaus können alle IVD jeweils noch für ein weiteres Jahr abverkauft werden.
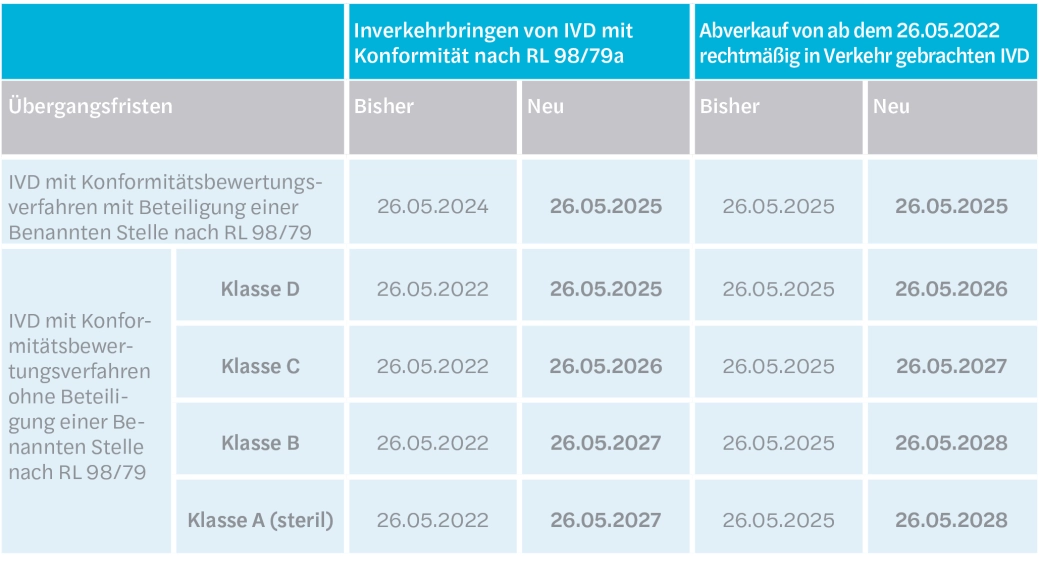
IVD-Hersteller, die von den Übergangsfristen profitieren, haben jedoch zu beachten, dass sie die Anforderungen der IVDR an die Überwachung nach dem Inverkehrbringen (Post-Market-Surveillance), die Marktüberwachung, die Vigilanz und die Registrierung von Wirtschaftsakteuren und Produkten bereits ab dem 26. Mai 2022 zu erfüllen haben.
Fazit:
Mit der IVDR werden die Anforderungen an die Qualität von Sars-CoV-2-Antigenschnelltests steigen. Zum einen sind zukünftig auch solche Testsysteme, die zum professionellen Gebrauch bestimmt sind, der unabhängigen Überprüfung durch eine Benannte Stelle unterworfen. Zum anderen müssen alle Sars-CoV-2-Antigenschnelltests unter der IVDR Anforderungen an die Mindestsensitivität erfüllen, um erfolgreich das Konformitätsbewertungsverfahren zu durchlaufen.
Trotz der Verlängerung der Übergangsfristen ist Herstellern von IVD schon jetzt empfohlen, die gestiegenen Regularien der IVDR mit ihren Prozessen und Dokumentationen abzugleichen und diese entsprechend anzupassen. Gerne unterstützen wir Sie im Hinblick auf alle regulatorischen Anforderungen rund um die IVDR. Darüber hinaus beraten wir Sie auch zu anderen mit IVD zusammenhängenden Fragestellungen, beispielsweise sozialversicherungs-, kostenerstattungs- und berufsrechtlicher Natur.
Haben Sie Fragen oder weiteren Informationsbedarf?
Autor*innen:
Julia Kleinschmidt
Tel: + 49 30 208 88 1037
Sebastian Retter
Tel: + 49 30 208 88 1043
Dies ist ein Beitrag aus unserem Healthcare-Newsletter 1-2022. Die gesamte Ausgabe finden Sie hier. Sie können diesen Newsletter auch abonnieren und erhalten die aktuelle Ausgabe direkt zum Erscheinungstermin.
Kontakt
